The audit that found nothing wrong.
An FDA auditor points to a deviation from six months ago: "Show me what happened here."
You pull up DEV-2024-047. The screen shows the batch step where it occurred, the equipment status at that moment, the operator's training record, and the investigation trail. The auditor clicks through to the CAPA. It closed three months ago after effectiveness verification. Zero recurrence in the following 50 batches, data attached.
The auditor moves on. No finding. No 483. This is what control looks like when you can prove it.
Your quality data lives in three systems, and auditors notice

Deviations in one system. Training records in another. Batch records in a third. To answer "was this operator trained when they executed this step during this deviation," someone assembles evidence from three sources. The answer arrives in hours, if the data exists at all.
The average 483 observation costs $200K-$500K to remediate. Warning letters cost $1-5M and take 12-18 months to resolve. Consent decrees can run into the hundreds of millions. None of this happens because someone did something wrong. It happens because someone couldn't prove they did something right.
This is how minor deviations become 483 observations. Not because of bad intent, but because the system couldn't prove control when asked.
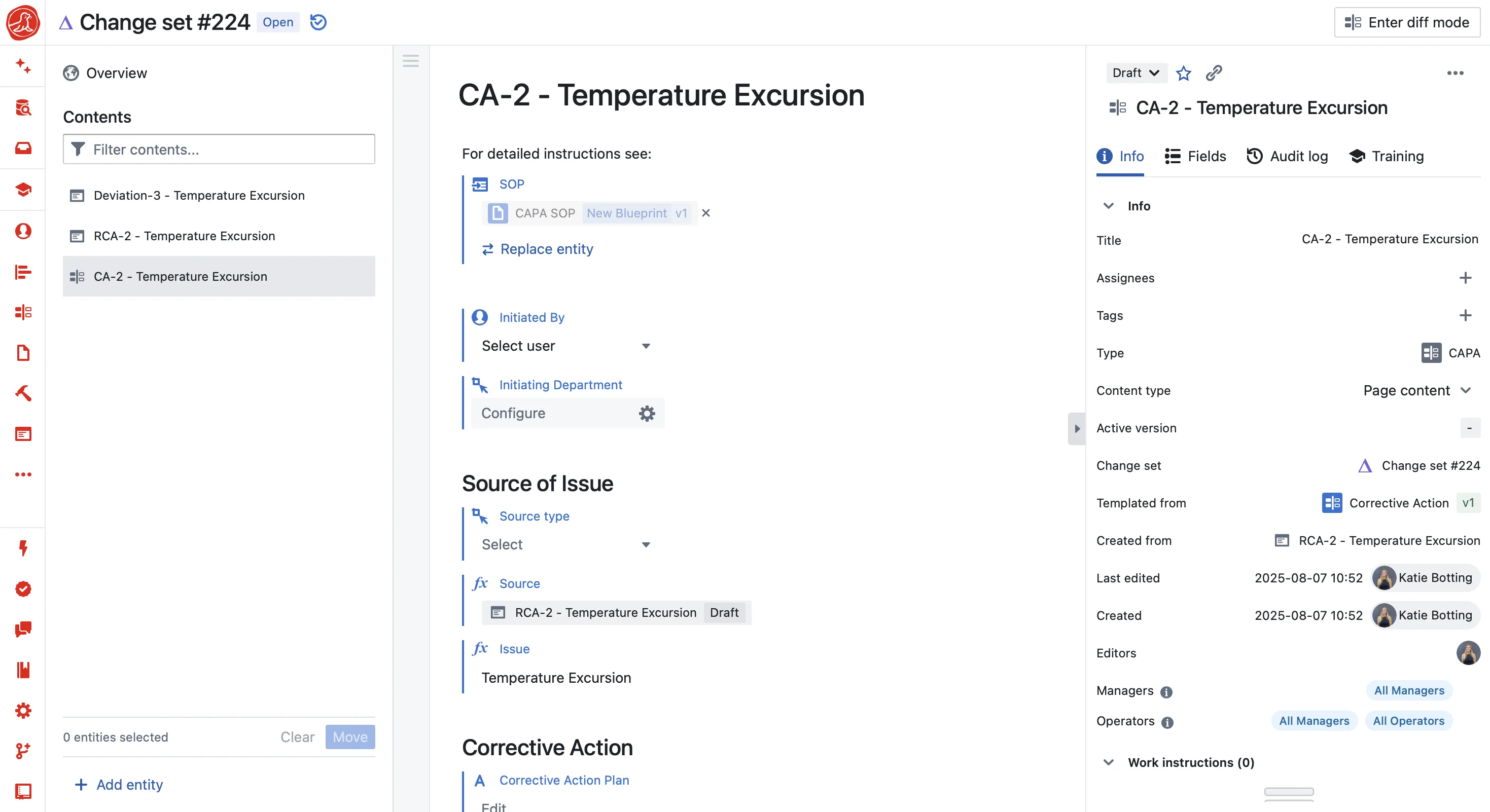
Every deviation arrives already linked to the batch, equipment, and operator

A deviation record in Seal contains the batch ID, equipment ID, process values, and operator training status at the time of the event. Because those records already exist in the same database. The connection isn't assembled after the fact. It's structural.
When an auditor asks to see a deviation, they see it linked to the specific batch step, equipment calibration status at that moment, and the operator's verified training. Complete chain of evidence, retrieved in seconds.
procedures are controlled objects; training is a gate, not a record
A procedure is a controlled object, not a file. Operators access only the current effective version. Superseded versions are archived and inaccessible for execution.
When Procedure-042 updates from version 2 to version 3, everyone trained on version 2 gets retraining assigned automatically. Until they complete training on version 3, they cannot execute tasks requiring that procedure. Training is a gate, not just a record. The system blocks execution before errors can occur.
21 CFR Part 11 compliance: immutable audit trails, meaning-based e-signatures (Author, Review, Approve), complete version history.
CAPAs can't close on paperwork — they close on verified effectiveness

Deviations capture in context. The record includes batch ID, equipment ID, and process values at the time of the event. Not because someone typed them in, but because that data already exists in the same system.
"Inadequate CAPA" appears on warning letters because organizations close CAPAs on paperwork, not proof. Seal requires effectiveness criteria upfront: "Zero recurrence in the next 100 batches." The CAPA cannot close until verification is complete and evidence is attached. If verification fails, the CAPA reopens automatically.
Quality threads connect across systems and never break

Quality threads connect across systems. An OOS result triggers investigation workflow immediately. Deviations link to batches. Batches link to materials. Materials link to suppliers. When an auditor follows a thread, it never breaks.
Annual Product Quality Reviews compile from live data. The data exists because it was captured during execution. There is no preparation phase. Audit readiness is the default state.
neil drafts your QMS change set
The hardest part of a QMS isn't using it. It's setting it up. Traditional implementations take months because consultants manually configure every deviation category, write every procedure template, and build every training matrix.
neil. Seal's AI. Proposes the configuration work. Tell neil you manufacture sterile injectables with three production lines and need to track deviations, CAPAs, and change control per FDA requirements. neil drafts deviation categories appropriate for injectable manufacturing, CAPA workflows with effectiveness verification gates, and change control classification matrices. Your quality team reviews and refines. Not builds from scratch.
neil drafts procedures from your process descriptions. Tell neil you need an aseptic gowning procedure per EU Annex 1. It generates a compliant draft with the required content structure. Need a training matrix? neil analyzes your procedures and role definitions and creates the curriculum: who needs training on what, in what sequence, with what assessment. Add a new procedure next quarter, and neil identifies who needs retraining based on their responsibilities.
neil also works inside your daily operations. A deviation opens. neil suggests root causes based on similar events in your history. A CAPA needs an action plan. neil drafts it from the investigation findings. A batch record needs review. neil highlights anomalies so QA focuses on exceptions. The AI that drafted your QMS helps you run it.
From overhead to prevention
Quality departments are often seen as cost centers. The team that creates paperwork. The department that says no. Every dollar spent on quality is a dollar that didn't go to R&D or sales.
This perception persists because traditional QMS systems are reactive. They document what went wrong. They don't prevent it. When the value of quality is "we wrote good reports about failures," quality looks like overhead.
Seal changes the equation. Training gates prevent untrained operators from touching critical steps. That's not documentation, that's prevention. Real-time deviation capture means problems surface before they become 483s. Effectiveness verification means CAPAs actually work. When quality prevents the $500K remediation instead of documenting it, quality becomes an asset.
The QA team that can answer any auditor question in seconds isn't overhead. The system that catches drift before excursion isn't bureaucracy. The training that blocks unqualified execution isn't paperwork.
Quality should be the department that protects the company. That requires a system designed for protection, not archaeology.
Migrate from paper, SharePoint, or a legacy electronic quality management system in weeks
Most customers are migrating from either paper, SharePoint, or a legacy eQMS that stopped working for them. All three paths are different.
If you're on paper or SharePoint, the path is straightforward: define your document hierarchy, import your current procedures, and start capturing quality events digitally. Training management can follow within weeks. You don't need everything configured to go live. Start with what you use most, expand from there.
If you're on a legacy eQMS, you have a choice. You can run parallel for a period while you migrate critical data, or you can archive the old system as read-only and start fresh. Most customers choose the second path. You don't need to migrate 10 years of closed CAPAs. You need to manage the next 10 years properly.
Either way, the timeline is weeks, not months. Document control typically takes 1-2 weeks to configure. Deviation and CAPA management adds another 1-2 weeks. Training integration follows. AI accelerates all of this. What used to require consultants now requires descriptions.