Paper batch records are holding you back.
Every deviation, every data integrity finding, every product recall traces back to the same root cause: humans copying numbers from one piece of paper to another. The paper batch record was a reasonable solution in 1970. Today, it's a liability consuming 20-30% of manufacturing time and causing error rates as high as 25%.
You know the cost. Months of release delays while QA reviews page after page. Millions in investigations when someone transposes a digit. Regulatory observations that question your data integrity. Yet most organizations cling to paper because they've been burned by failed digital transformations. Companies who bought an "Electronic Batch Record" system that promised to eliminate paper and got a PDF on a screen instead. Same rigid workflows, same manual transcription, same review process, just on an iPad instead of a clipboard.

The fundamental problem is that these systems digitized the wrong thing. They digitized the paper form instead of the manufacturing process. The form is just a documentation artifact. The process is what matters.
Your MES should evolve with your process, not freeze around it
Legacy MES implementations assume your process stabilizes. You validate once, freeze the configuration, and operate in that frozen state until something forces a change. At which point you pay consultants to unfreeze it. For a biotech in Phase 2, a C> company running autologous therapies, or any organization whose process is still moving, that model doesn't fit.
Seal is architected around the opposite assumption: your process evolves continuously, and your MES should evolve with it. CPP range tightening based on new PPQ data flows through change control in days, not a year of re-IQ/OQ/PQ. A new site inherits the Platform Process and binds site-specific configuration. A new product variant reuses existing unit operations rather than requiring a new validation package. Change control is real. Every change is explicit, tracked, approved. But the scope is the actual change, not the entire system.
For C> specifically, every patient batch is a variant of the process. Batches of one, chain-of-custody from apheresis to infusion, per-patient lot genealogy. Paper batch records cannot handle this operationally. Legacy MES built for mass production cannot model it semantically. Seal handles it because the platform is configured. Not hard-coded. To your process, and AI configures the per-patient variants as they're needed.
For early-stage biotech, the same architecture matters for a different reason: you cannot afford an MES you will outgrow. Seal starts with your first GMP batch and scales to commercial without rip-and-replace. Your first ten batches, your first hundred, your first thousand. Same platform, same data model, same audit trail, same vendor. The MES you pick at Phase 2 is the MES you run at commercial launch, not a stepping-stone you'll throw away.

Process as data, compliance by enforcement
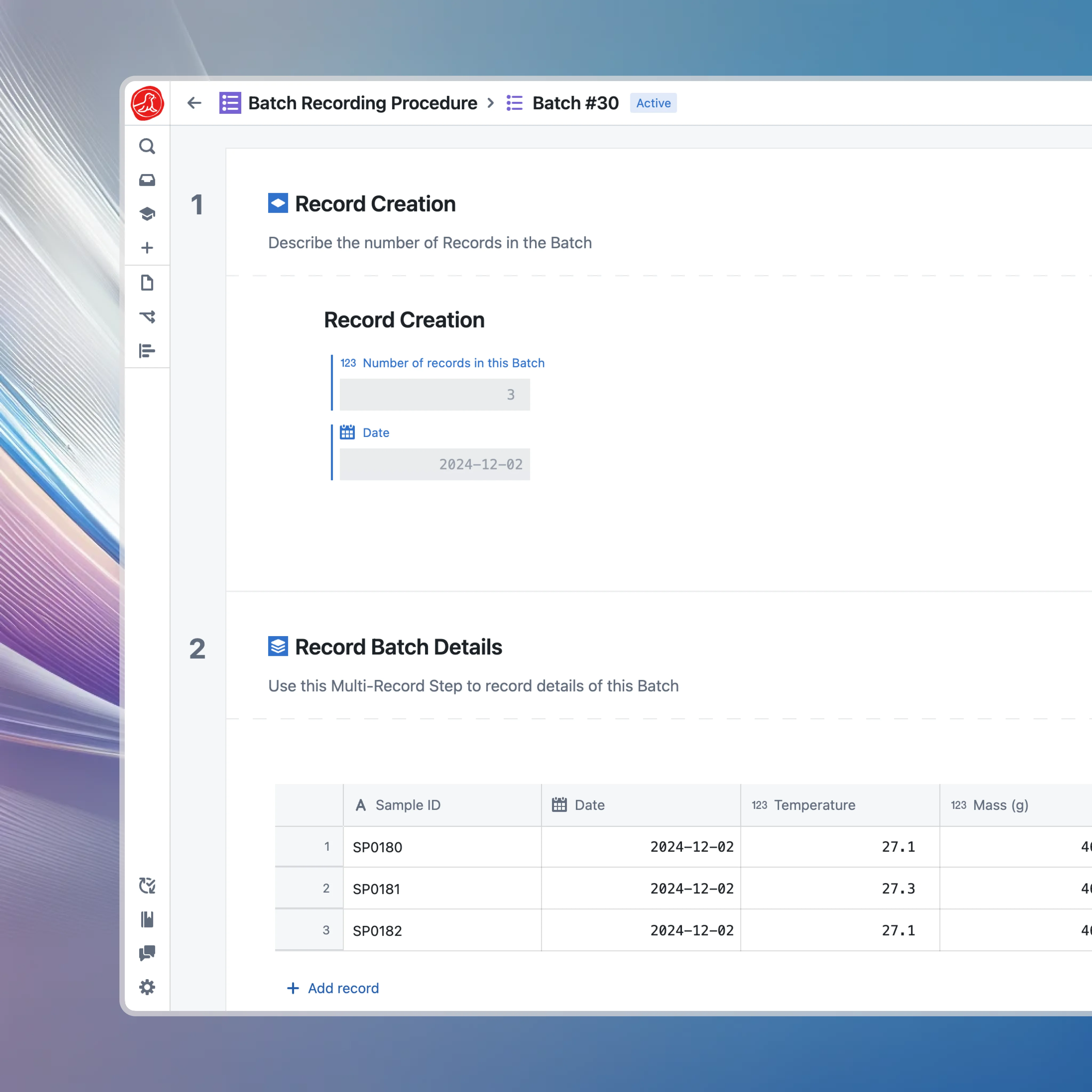
Seal treats your manufacturing process as structured data. A batch record isn't a form to fill out. It's a workflow to execute. Each step knows what inputs it needs, what outputs it produces, what equipment it uses, and what parameters must be within range. When an operator executes a step, they're not filling in a blank field. They're interacting with the process itself. The system knows what should happen and guides them through it. Parameters are captured from connected equipment, not typed in. Material lot numbers are scanned, not written. Calculations are performed automatically, not done on calculators and transcribed.
This structure enables something paper can never do: enforcement. An operator cannot proceed if a value is out of specification. They cannot use expired materials. They cannot skip a required verification. The compliance controls are built into the process, not enforced through review after the fact.

When compliance is enforced at the point of execution, the review process transforms. You're not reading 500 pages hoping to catch an error. You're reviewing the three exceptions that occurred during the batch: the times someone had to override a warning, the parameters that approached limits, the decisions that required judgment. Review time drops from weeks to hours, and review becomes meaningful. Instead of checking that data was transcribed correctly, reviewers focus on the decisions that actually matter.
The downstream effect is on cash flow, not just operations. Every day a batch sits in release review is a day of working capital frozen in inventory. In a commercial operation manufacturing twelve batches a month, a three-week review cycle means roughly thirty-six batch-weeks of inventory frozen at any given time. Collapse that to three days and you free working capital proportionally. For a CDMO running client campaigns, the same shift changes what you can fit into a quarter: more batches released inside the window where the client's downstream activities expect them.

The shape of the curve is what changes. Traditional release is a long queue with unpredictable endings. You can't forecast release date on a batch until QA is halfway through its review, because you don't know how many deviations are buried in the paperwork. Seal releases are predictable because the deviations that would have been discovered in paper review surface at the moment they occur, during execution. By the time QA starts review, they already know what happened and what needs adjudication.
Equipment data and material lots flow into the batch record automatically
Manufacturing equipment generates enormous amounts of data that traditionally gets ignored or manually transcribed. A bioreactor logs temperature every second. Tens of thousands of data points per batch. Yet paper-based operations reduce this to "temperature maintained within range" with a single checkmark. Seal integrates directly with bioreactors, fillers, autoclaves, balances, and other equipment to capture process data automatically. See instrument connectivity for how the connection works, including floors that run offline.

Temperature profiles, pressure readings, weight measurements, timing data. All of it flows into the batch record without anyone typing anything. The data that enters your quality record is the data that came from the equipment, with complete traceability and no transcription errors. When an auditor asks "what was the temperature at 14:32:07?", you have the answer. Because the bioreactor told you, not because someone wrote it down.
Material dispensing is where many manufacturing errors originate. The wrong material, the wrong lot, the wrong amount. Any of these can destroy a batch. Operators scan material barcodes and the system verifies it's the correct material and lot. Balances connect directly so weights are captured automatically. The system calculates required amounts based on the batch size and enforces tolerances. If something is wrong, the operator knows immediately. Not hours or days later when someone reviews the paperwork.

When an operator scans a material barcode, the system verifies five things in milliseconds: Is this the right material? Is this the right lot? Is it released for GMP use? Is there enough quantity? Has it expired? If any check fails, the operator cannot proceed. The system tells them exactly what's wrong. When materials pass verification, the system captures the lot number in the batch record and decrements inventory automatically. No separate inventory transaction. No reconciliation at end of day. The batch record and inventory system stay synchronized because they're the same system.
Unqualified operators can't start steps; uncleaned equipment can't start batches
An untrained operator executing a critical step is a deviation waiting to happen. In paper-based systems, you discover the problem weeks later during batch review: "Wait, did Martinez complete their aseptic technique requalification before this fill?" With Seal, you never get to that question. Because an unqualified operator cannot execute the step in the first place.
Each step in your batch record can require specific training curricula. "Aseptic Fill Step 3" might require qualification on the specific filling line, current gowning certification, and completion of the aseptic technique procedure. The operator scans their badge. In milliseconds, the system checks their training record against all requirements. If they're qualified, they proceed. If they're not, they can't. The system won't let them start the step. When training expires, the system knows instantly. An operator who was qualified yesterday but whose annual recertification lapsed at midnight cannot execute those steps today. Compliance is continuous, not point-in-time.
Between batches, equipment must be cleaned. Between products, lines must be cleared. Seal treats cleaning as a first-class workflow, not a checkbox on a paper form. Cleaning records link to three things: the equipment cleaned, the batch that dirtied it, and the batch that will use it next. Swab test results flow in from LIMS. If the rinse water conductivity is out of spec, you know before the next batch starts, not after it's contaminated. Hold time calculations enforce your validated cleaning hold times. If your validation says equipment must be used within 72 hours of cleaning, the system knows. And won't let you start a batch on equipment cleaned 73 hours ago.
Line clearance for product changeover follows a structured checklist. The system won't allow the new product to start until every clearance item is complete. This isn't paperwork. It's a gate. The contamination events that destroy batches and careers happen when someone starts a new product before the line is truly clear.
The downstream effect on training programs is often underestimated when evaluating an MES. When qualification is enforced at the point of execution, the training team discovers within days which curricula are actually required for which steps. Because steps with missing qualifications block execution until curricula are assigned. The same structural link tells you which operators are approaching expiration and who needs retraining scheduled before they're blocked from shift. The training program moves from "send reminders and hope" to "here are the seventeen qualifications expiring in the next fourteen days across three shifts."
The deviations this prevents are specific and familiar. The aseptic fill where the operator's requalification lapsed the day before. The weigh-and-dispense where the balance certification expired six weeks ago and nobody noticed. The cleaning step where the operator was qualified on a different line. The batch sign-off where the person signing was not trained on the step they signed for. Each of these is a Warning Letter waiting to happen in a paper-based shop; each of these is impossible in Seal because the system checks before permitting.

Sampling and hold times are enforced inside the batch record, not alongside it
Manufacturing isn't fire-and-forget. Critical steps require in-process checks: pH verification before proceeding, osmolality confirmation, bioburden samples, appearance checks. The sampling workflow is where paper-based operations fall apart hardest. The operator takes a sample, writes the batch and step number on a label, carries it to QC, the QC team logs it into their own system, tests it, writes the result back to the operator or supervisor, who writes it into the paper batch record. Every one of those handoffs is an opportunity for error: wrong batch number written on the label, wrong step referenced, result lost in email, transcription off by a decimal. Most batch records that fail QA review don't fail because the manufacturing went wrong. They fail because the sampling paperwork and the manufacturing paperwork don't agree on what happened when.
Seal manages the sampling workflow alongside the manufacturing workflow, ensuring checks happen when they should and results link to exactly the right step.

At Step 12, the batch record calls for a pH check. The operator takes the sample and scans the sample container. The system creates a sample record in LIMS. With the batch number, step number, timestamp, and operator ID already populated. And pauses the batch. The batch cannot proceed to Step 13 until LIMS returns the result. If pH is within range, the result populates in the batch record and the batch proceeds automatically. If it's out of range, the system flags the exception and routes to your defined escalation path.
Because LIMS and MES live on the same platform, the sample record is not "created in a separate system via an integration." It is the same record. The batch knows the sample is pending because the record itself carries its status; QC knows which batch and step the sample belongs to because those fields are the record. When the result is committed, the batch sees it immediately. Not after an overnight sync, not after someone notices the result in LIMS and walks it over to manufacturing. The "integration" that typically causes the most deviations in a hybrid MES/LIMS stack simply doesn't exist here because there are no integrations to fail.
Hold times are enforced automatically. If your process validation established that Step 15 must start within 4 hours of Step 12 completing, the system tracks this from the moment Step 12 completes. At 3 hours, warnings appear. At 3.5 hours, the warning escalates to supervisors. At 4 hours, if Step 15 hasn't started, the batch cannot proceed without documented justification and approval. This isn't nagging; it's enforcement of the constraints your validation established.
Each hold time constraint is linked to the validation study or procedure that established it. When an operator bumps into a hold-time wall, the system surfaces not just the warning but the rationale: "Step 15 must start within 4h of Step 12 per study PV-2024-03, which demonstrated protein aggregation above 4h hold." Operators and supervisors don't have to hunt for the reason the constraint exists. The constraint carries its justification. This closes the most common path to avoidable deviations: an operator or supervisor overriding a constraint they don't understand.
Every batch is fully traceable, in real time and after the fact
Stop guessing where your batches are. Seal turns your facility into a glass box, providing real-time visibility into every aspect of manufacturing operations.

You know exactly which step every batch is at. You know which rooms and equipment are occupied and when they'll be available. You can see a batch falling behind schedule and intervene before it becomes a problem. Every batch has a story, and with Seal, you can tell it.

Click any finished product and see its complete genealogy: every raw material lot that went into it, every piece of equipment it touched, every operator who worked on it, every environmental condition it experienced, every test result from QC. Forward traceability works too: click any raw material lot and see every batch that used it, every product that contains it. When a supplier issues a recall notice at 3pm, you know by 3:05pm which batches are affected. When a customer reports a quality issue, you can trace back to exact conditions. Not "around January" but "Lot FP-2024-041, manufactured 2024-01-15, using API lot RM-892 from Acme, mixed on equipment MX-001 for 2 hours at 200 RPM." This isn't archaeology. It's a query.
MES, LIMS, and QMS share the same records, with compliance built into the architecture
Because your MES lives on the same platform as your LIMS and QMS, the handoffs that create delays in traditional systems don't exist. QC results from the LIMS appear directly in the batch record as testing completes. No waiting for someone to email a spreadsheet. Deviations open in the QMS directly from the manufacturing step that triggered them, with full context captured automatically. For batch release, all the information needed to make a release decision is in one place.

The alternative architecture. A standalone MES from one vendor, integrated with a standalone LIMS from another vendor, integrated with a standalone QMS from a third vendor. Is the single largest source of pain in life-sciences software today. Every integration has its own data model mismatch. Every sync has its own failure modes. Every system has its own change-control workflow, so a CPP range tightening proposed in the LIMS has to be separately proposed and approved in the MES. Every system has its own audit trail, so when an inspector asks why a parameter was set where it was set, the answer lives across three systems that don't agree on what a "parameter" is. The enterprise IT team becomes the reconciliation engine; it's a job that never finishes.
Seal removes the category of problem. A unit operation in the ELN is the same record that executes in the MBR. A sample specification in the LIMS is the same specification that the MBR's IPC step references. A deviation raised in manufacturing is the same deviation that QMS tracks. Not copies, not synced, not integrated. One record, one change history, one audit trail.
21 CFR Part 11 and EU Annex 11 compliance isn't a feature to add. It's an architecture to build on. Every signature requires two-component authentication with re-authentication at the moment of signing. The signature captures who signed, when they signed, and what they were signing. The exact record state at signature time. The meaning of the signature is explicit: "I performed this step" differs from "I verified this step" differs from "I approved this batch." Signatures are linked to training records and role assignments. An operator can sign that they performed a step but cannot sign that they verified it unless they have the verifier role.
The audit trail is immutable by design. ALCOA+ compliant from architecture, not policy. Every action in the system is logged with user, server timestamp, and before/after values. You cannot delete audit trail entries. You cannot modify them. There is no "admin override" that bypasses the audit trail. Data is encrypted in transit and at rest, with SOC 2 Type II certification and records retained for a minimum of three years beyond your subscription.
Start with one unit operation, validate it, then expand
Timing matters. If you're in early-stage R&D or Phase 1 with infrequent batches and constantly changing processes, an MES might constrain you. Stick to a flexible ELN and paper until your process stabilizes. If you're entering Phase 3 or commercial launch, implementing an MES during a pivotal trial is high-risk. The sweet spot is Phase 2 or early Phase 3: your process is stabilizing, your volume is increasing, and you need to lock down compliance before commercialization.
You've seen the legacy MES implementations. Eighteen-month timelines that stretch to three years, budgets that double, go-lives pushed quarter after quarter. Seal works differently. You start by digitizing a single unit operation. Weigh and dispense, perhaps, or a critical filling step. Within days, not months, your operators execute that step digitally. Your QA team sees what review by exception actually looks like. You validate one workflow, not an entire system. You see the value before you've bet the company on it.
Converting paper batch records to digital workflows shouldn't mean re-typing every step. Drop your existing master batch records. Word documents, PDFs, even scanned paper. And AI extracts the structure. A 50-page paper MBR becomes a structured digital workflow in hours, not weeks. AI identifies unit operations, process parameters, material requirements, equipment specifications, and verification steps. Drop your process validation reports and AI extracts the proven acceptable ranges. The pattern is consistent: AI extracts, you review the changeset, approved data enters the system. Your process experts validate the extraction. They're not typing it.
The phased approach matters at the human level too. Most MES failures are not technology failures. They're change-management failures. Operators who've spent years writing on paper don't adopt a new system because the system is better; they adopt it because one step at a time feels possible. Seal's phased rollout is designed to respect that. A single unit operation goes digital in week one. Your operators execute that step on a tablet while the rest of the batch still uses paper. They form opinions based on that one step: does the tablet work with gloves, does the barcode scanner catch on the first try, does the prompt for the second witness signature show up where it should. You hear what's working and what isn't, adjust, and move the second unit operation when operators are asking for it rather than resisting it. By the time the full batch record is digital, the operators have already been using Seal for months. It isn't a change, it's an extension.

The validation path reflects the same phased model. You validate one workflow at a time. A single unit operation, a single MBR section, a single site. Each validation is scoped to what actually changed, which means the validation effort tracks the implementation effort rather than running as a separate twelve-month project. The end state is a validated MES; the path there doesn't require a frozen twelve-month implementation window during which nothing else can change.
Every batch your team runs today generates data that could drive faster release, fewer deviations, and better process understanding. Paper buries that data in binders. Legacy EBR systems trap it in rigid forms. Seal liberates it. Your process, digitized. Your compliance, enforced. Your data, accessible. Your team, focused on what matters.